

Childhood acute myeloid leukemia (AML) is an aggressive myeloid malignancy characterized by mutational and cytogenetic abnormalities. Chromosomal rearrangements involving the NUP98 gene have come to light for its' significant impacts on outcome and response to treatment. NUP98-rearranged (NUP98-R) AML includes NUP98-NSD1, NUP98-KDM5A, and various less common NUP98 fusion partners - such as HOX, SET, and Bromodomain genes. NUP98-R account for 6.8% of patients across Children's Oncology Group studies CCG2961, AAML03P1, AAML0531, and AAML1031. The majority are NUP98-NSD1 (4.6%), then NUP98-KDM5A (1.4%), and the various partners, termed NUP98-X (0.83%). However, the biological implications of NUP98-X have yet to be investigated. We define transcriptional clusters and report transcriptional profiling results to reveal similarities and differences between the diverse NUP98-R fusions.
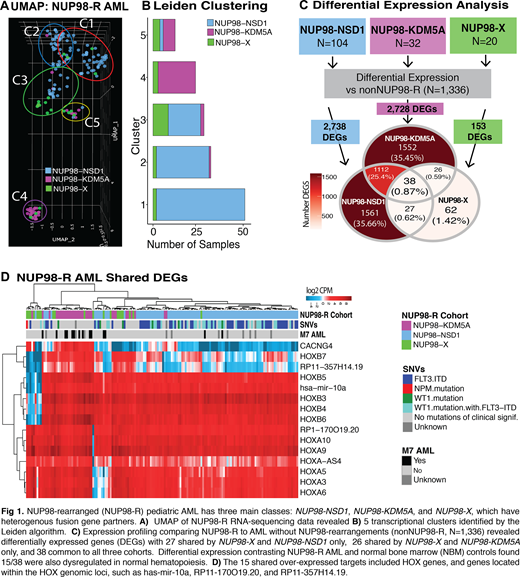
RNA sequencing was completed for 1,492 pediatric AML patients and 68 healthy bone marrow controls (NBM). The algorithms STARfusion, TransAbyss, and CICERO detected 156 NUP98-R from RNA seq: NUP98-NSD1 (N=104), NUP98-KDM5A (N=32), and NUP98-X (N=20). NUP98-X encompassed 13 unique fusion partners and 45% (9/20) had a homeobox gene partner - HOXA9 (N=4), HOXD13 (N=3), HOXA13 (N=1), and PRRX1 (N=1). Unsupervised clustering via uniform manifold approximation and projection (UMAP) with input genes selected by jackstraw PCA revealed NUP98-X cluster more closely with NUP98-NSD1, but their trajectory is dispersed within NUP98-NSD1 and NUP98-KDM5A, indicating a potential hybrid transcriptional profile (Fig 1 A). NUP98-NSD1 clustered remotely from the majority of NUP98-KDM5A, highlighting their unique transcriptional profiles despite sharing NUP98-R. Five transcriptional clusters were identified by the Leiden algorithm, followed by selection of genes significantly associated with cluster assignment. Cluster marker genes were filtered to be unique for each cluster prior gene-set enrichment analysis (Fig 1 B). Cluster C3 encompassed the largest proportion of NUP98-X, with 7 homeobox and both PHF23 cases (9/20, 45%), but also comprised of 19 NUP98-NSD1 and 2 NUP98-KDM5A. Importantly, C3 was highly associated with the expression of long non-coding RNAs, Z69666.2, MT1XP1, and RP11-455O6.2 (≥ 37% specificity, FDR < 0.001), and pathways in enriched in oncogenic microRNAs (FDR < 0.001), suggesting a non-coding signature may define this group. NUP98-NSD1 divided into two primary clusters, C1 (52/104) and C2 (31/104), differentiated by the expression of mannose receptor genes MCR1/ MRC1L1, which detect products of the lysosome pathway, significantly activated in C1 (FDR < 0.001). NUP98-KDM5A also segregated into two clusters, C4 (N=22/32) and C5 (N=7/32) based on an FAB M7/AMKL signature, with 78.6% of M7 NUP98-R located in C4 (p=0.003). Whereas, C5 cases were non-M7 and uniquely enriched in the regulation of stem cells pathway (FDR = 0.005); activation was driven by the expression of POU5F1, BMP4, WNT7, and WNT10A/B.
Transcriptional profiling of NUP98-R cohorts, independently compared to other AML (N=1,336), found 27 upregulated differentially expressed genes (DEGs) shared between NUP98-X and NUP98-NSD1, including MYCN oncogene, homeobox PBX3, and DNA methyltransferase DNMT3B; on the other hand, shared overexpression of MLLT3, homeobox IRX3, and CD79a characterized similarities between NUP98-X and NUP98-KDM5A (Fig 1C) and may contribute to the hybrid expression profile observed by UMAP clustering. There were 38 DEGs shared between all NUP98-R cohorts. Differential expression analysis comparing NUP98-R cohorts vs NBM (N=68) showed that 22/38 shared DEGs were dysregulated in normal hematopoiesis and 15/38 genes were concordantly overexpressed in NUP98-R. The minimal set of 15 genes strongly implicates dysregulation at the HOX locus; these targets include hsa-mir-10a, whose genomic locus is within the HOXB cluster, CACNG4 located on chr17q, the same chromosome arm as HOXB, and the remaining shared targets were HOXA/B genes (Fig 1D).
In summary, we found that NUP98-X are dispersed between NUP98-NSD1 and NUP98-KDM5A by transcriptional clustering. Clustering revealed significant pathways and marker genes that may contribute to segregation of NUP98-R cohorts, and DEGs that contribute in part to their hybrid transcriptional profile, including HOX genes, PBX3, and IRX3.
No relevant conflicts of interest to declare.

